


 (This photo was taken from CBS.com)
(This photo was taken from CBS.com)
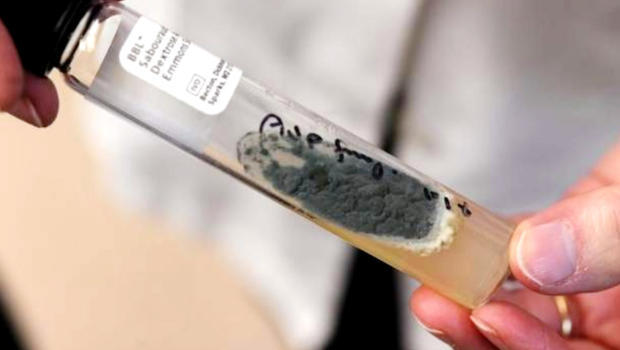
Last night “60 Minutes” did a segment on “Lethal medicine linked to meningitis outbreak.” The New England Compounding Center was producing a steroid that helps to relieve chronic pain in patients’ backs and joints. This steroid has lead to 50 deaths and other significant problems for 700 innocent patients. This is all due to the lack of drug safety. The steroid is contaminated with fungus, which leads to patients contracting meningitis. This meningitis can go to a patient’s bones, nerves and even brain. When the meningitis gets to the brain there is a very low percentage of patients who survive. This lethal medicine has caused many issues for those who have used it; many of these problems cannot be cured and the lives taken cannot be replaced.
The New England Compounding Center in Massachusetts got greedy and “sloppy,” said a former employee who spoke on “60 Minutes.” They were trying to produce mass amounts of drugs while reaping the significant benefits. Therefore, the drug preparation became unsterile. There was mold found in the Clean Room One at the NECC 12 times in three years. A supervisor was warned and according to the employee who was interviewed on “60 Minutes,” the supervisor just shrugged and didn’t say anything about the issue.
According to the “60 Minutes” segment, “the New England Compounding Center was what’s known as a compounding pharmacy. By law, compounding pharmacies are not allowed to manufacture pharmaceuticals for the mass market. That would require the oversight of the FDA. Instead, the states are licensing compounding pharmacies to make drugs for individuals. For example, a doctor might order a liquid form of a medication for a patient who can’t swallow a pill. Compounding pharmacies are bound by one rule: they must have a prescription for each individual patient.”
The NECC was shipping tens of thousands steroids out of the Clean Room One. It was stated that the 3,000 clients that NECC sold to were part of the fraud. The Clean Room One was shown on “60 Minutes” for the first time for the public.
There are many concerning issues for patients. One such concern is that there is no cure for those patients who are infected by the fungus. To date, no doctor has said the fungus is completely gone from an infected patient’s body. It is a constant worry and according to patients who are infected, the treatment is very rough. The other concerning issue is that as patients, we do not know what drugs are approved by the Food and Drug Administration. We simply trust the care of the producers to have safety precautions and make drugs properly. Clearly, those producers cannot always be trusted.
It is still unclear on how the drug was contaminated. What we do know is that there is no cure and the consequences are severe. When the FDA went into the NECC for a safety and cleanliness check, they found 50 samples of the medicine and they were all contaminated with fungus. The company is now in bankruptcy and has faced many trials. However, a life of a loved one cannot be replaced. If you or a loved one has experienced a situation similar to this one please give us a call. No one deserves this kind of treatment.
 As a patient, being a health advocate is important. As our world progresses so does technology and the advancement in the medical field. Companies are making products so quickly, so the product can be sold in stores. Often products have not been tested to ensure the product works correctly. This therefore leads to severe consequences. As patients and citizens we trust doctors and hospitals to care for us in a proper and safe manner. We also trust companies to produce products that are not harmful and work correctly, although more times than not this does not happen. From simple products that you receive from the convenience store to products that could save your life, need to work properly.
As a patient, being a health advocate is important. As our world progresses so does technology and the advancement in the medical field. Companies are making products so quickly, so the product can be sold in stores. Often products have not been tested to ensure the product works correctly. This therefore leads to severe consequences. As patients and citizens we trust doctors and hospitals to care for us in a proper and safe manner. We also trust companies to produce products that are not harmful and work correctly, although more times than not this does not happen. From simple products that you receive from the convenience store to products that could save your life, need to work properly. .While the benefits of these new methods of treatment are undeniable, a side effect of these drugs is pancreatic cancer.
.While the benefits of these new methods of treatment are undeniable, a side effect of these drugs is pancreatic cancer.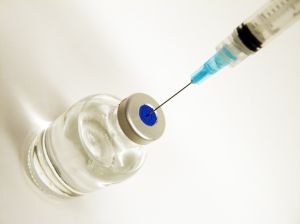 A pain shot made in a Tennessee pharmacy may be contaminated and could be making people sick. This is very similar to the meningitis outbreak that happened last year. The results of this outbreak are less severe than the outbreak of the meningitis illnesses, but there are infections at the site of where the pain shot was inserted. These infections are skin and soft tissue of unclear etiology following the injection. This is leading to many patients having abscesses. An abscess occurs when white blood cells move through the walls of the blood vessels to the site of infection and collect in the area of damaged tissue. The result of this is the presence of pus. Pus is the buildup of fluid, living and dead white blood cells, dead tissue, and bacteria or other foreign substance. Abscesses can show up anywhere on the body. The most common sites are in your armpits, areas around your anus and vagina, the base of your spine, around a tooth, and in your groin. However if you have received a pain shot, abscesses would form at the site of the shot.
A pain shot made in a Tennessee pharmacy may be contaminated and could be making people sick. This is very similar to the meningitis outbreak that happened last year. The results of this outbreak are less severe than the outbreak of the meningitis illnesses, but there are infections at the site of where the pain shot was inserted. These infections are skin and soft tissue of unclear etiology following the injection. This is leading to many patients having abscesses. An abscess occurs when white blood cells move through the walls of the blood vessels to the site of infection and collect in the area of damaged tissue. The result of this is the presence of pus. Pus is the buildup of fluid, living and dead white blood cells, dead tissue, and bacteria or other foreign substance. Abscesses can show up anywhere on the body. The most common sites are in your armpits, areas around your anus and vagina, the base of your spine, around a tooth, and in your groin. However if you have received a pain shot, abscesses would form at the site of the shot.